





Chronic inflammation evoked by pathogenic stimulus during carcinogenesis
Abstract
A pathogenic (biological or chemical) stimulus is the earliest information received by a cell that can result in the disruption of homeostasis with consequent development of disease. Chronic inflammation involves many cell types with numerous cytokines and signaling pathways, the release of different components by the cells, and the crosstalk provoked by such stimuli involving subclinical chronic inflammation and is mechanistically manifold. Exosomes secrete chemicals that trigger the epithelium to produce exosome-like nanoparticles promoting chronic inflammation. Small molecules, together with various cytokines, selectively target signaling pathways inducing crosstalk that suppress apoptosis. 16S rRNA gene sequencing has become routine to provide information on the composition and abundance of bacteria found in human tissues and in reservoirs. The deregulation of autophagy with chronic stimulation of inflammation is an early phenomenon in carcinogenesis. The disruption of cell–cell integrity enables transcellular CagA migration and triggers deregulation of autophagy with the net result being chronic inflammation. The complex and insidious nature of chronic inflammation can be seen both inside and outside the cell and even with intracellular nuclear fragments such as chromatin, which itself can elicit a chronic inflammatory response within the cytoplasm and affect autophagy. The ultimate result of unresolved chronic inflammation is fibrosis, a step before tissue remodeling results in the formation of a precancerous niche (PCN). Various pathogenic stimuli associated with different neoplasms result in persistent inflammation. This ongoing disruption of homeostasis in the micromilieu of cells, tissues, and organs is an essential preamble to carcinogenesis and occurs early in that process.
Key words: Adenoma / Adhesion / Akt / ALOX / Apoptosis / Aquaporin / Autophagy / Bacterium / BIM / Blastoma / Cancer / Carcinoma / Carcinogenesis / CCC / Cdc42 / Cdk2 / Cholangiocellular carcinoma / Crohn's disease / Chronic inflammation / Colitis / Colorectal cancer / COX / Cyclin / Cyclooxygenase / CYP / Cytochrome P450 / Cytokine / CXCR4 / E2F4/5 / E-cadherin / Eicosanoide / EBV / Epstein–Barr virus / ERK / ETE / Fibroblast / Fibrosis / Fluke / FOXO3a / Gastric cancer / Gastritis / Glycocalyx / HBV / HCV / Helicobacter pylori / Hepatitis B virus / Hepatitis C virus / HETE / Homeostasis / HCC / HIV / HPV / HSV / Human herpes virus / Human papilloma virus / IBD / ICAM / IDO / IL / IL-β1, Interleukin / Inflammation / Leukemia / Lipoxygenase / LTA4 / LTB4 / LTC4 / LTD4 / LTE4 / Liver cancer / LOX / LOXL3 / Lymphoma / Lysyl oxidase / MAPK / MDA / Metalloproteinase / MMP / Mutation / NF-κB / AP1 / API2 / PCN / PGD2 / PGG2 / PGH2 / PGFF2a / Phagocytes / PI3K / Polyp / Precancerous niche / Prostate cancer / PUMA / Rac1 / RNS / ROS / Sarcoma / SPhK / S1P / S1PR3 / Simvastatin / SK2 / SOX / Tissue / TGF / TNF / TOR / TXA2 / VCAM / Virus / VZV
Introduction
Inflammation associated with cancer dates back to the British physician Sir Percival Pott (1714–1788), who reported chronic inflammation in testicular cancer in chimney sweeps in 1755 [1]. In 1829, William Edmonds Horner (1793–1853) described how chronic inflammation follows unresolved acute inflammation [2]. The French physiologist, Henri Dutrochet (1776–1847), reported how cells migrate to sites of inflammation [3]. In 1843, an experimental injury-induced diapedesis was shown by William Addison (1802–1881) [4], which was followed by the British physiologist, August Volney Waller (1816–1870) in 1846, who observed colorless blood cells in intact capillaries within frog tongue and mesentery [5]. In 1857, Brown reported acute (and chronic) inflammation induced by the topical application of arsenic in a patient [6].
Rudolf Virchow reported leukocytes in cancer tissues [7] and his pupil, the German-Jewish pathologist, Julius Friedrich Cohnheim (1839–1884), showed that colorless blood corpuscules migrated from vessels into inflammatory tissue and named these cells Eiterkörperchen (pus-corpuscles) [8,9], which resulted into the so-called migration theory and introduced a renaissance in the theory of inflammation. In contrast to his teacher Rudolf Virchow, Cohnheim found that “the leukocytes found in inflammation are predominantly derived from the blood by diapedesis through the capillary walls” (reviewed in Ref. [10]).
In 1868, the British Surgeon Thomas Bryant reported 10 cases of chronic inflammation in female breast cancer patients [11]. His fifth case was a 48-year-old female who presented with a history of earlier abscess and chronic inflammation of her right breast, which he was suspected of having cancer. Treatment against inflammation with tonic was successful but the patient developed contralateral breast cancer a year later. In 1891, Frederic S. Eve reported a 48-year male with chronic inflammation and tongue cancer with “chronic superficial glossitis for years” and a sore tongue for some 10 years [12].
Cohnheim's leukocytes migration was proven by the Virchow's pupil, Julius Arnold (1835–1915) in 1873 [13–15]. In 1883, the Russian Zoologist Elias Metschnikoff (1845–1916, also Ilya Ilyich Mechnikov from the Russian: Илья Ильич Мечников) observed phagocytic cells surrounding rose thorns stuck into starfish larvae as a host defense [16,17]: the phagocyte doctrine was born after Mechnikov's friend, Karl Claus, coined the term phagocytes. Metschnikoff was awarded the Nobel Prize in 1908. Various cell types during those early days had to be discovered, which may explain the difficulty to diagnose chronic inflammation-induced breast cancer [11].
Today we know that inflammation is a complex mechanism as any kind of stimulus appears to induce inflammation. The following is divided into pathogenic stimulus including viruses, bacteria, and fluke, followed by the newly described aquaporins (APQ) and autophagy followed by a section on chronic inflammation.
Pathogenic stimulus
A pathogenic (chemical or biological) stimulus is the earliest information a cell receives that can trigger the six sequential steps that lead to cancer [18,19]. The role of chemical carcinogens in the tumor microenvironment and carcinogenesis had earlier been reviewed. There is no published study wherein chemical carcinogens have been shown to induce somatic mutations and those mutations have led directly to the onset of cancers.
In fact, most studies on chemical carcinogenesis require a long latency period between repeated exposures to the carcinogen and the onset of cancer [20]. The reason for the latency period is the necessity of chronic inflammation and fibrosis with their associated changes in signaling that leads to the onset of cancer. It seems rather logical to combine results of observations in biology that the onset of cancer involves a pathogenic stimulus, followed (if unresolved) by chronic inflammation, fibrosis, and a disruption of homeostasis such that cancer can result over decades in a subset of the exposed population [18,19].
The fact that somatic mutations are detected at certain time points, post-exposure to chemical carcinogens [21,22], merely suggests that such mutations are epiphenomena or incidental to carcinogenesis and not necessarily causal to the onset of cancer as explained elsewhere [18,19,23].
For example, chronic exposure to certain asbestos fibers results in pleural mesothelioma but with no mutations [24]. Carbon tetrachloride causes inflammation, fibrosis of the liver, and later hepatic carcinoma [25]. Hepatitis C in humans causes chronic inflammation with liver fibrosis, cirrhosis, and some 30% of patients with cirrhosis go on to develop hepatocellular carcinoma (HCC) with no mutations [26]. This is also true in the case of Human Papilloma virus (HPV) and cervical or oropharyngeal cancers with chronic inflammation but no somatic mutations [27–30]. Although we all acknowledge that chronic cigarette smoking raises the risk of lung cancer, the fact that only about 10% of chronic smokers develop lung cancer [31] suggests that a pathogenic stimulus is not sufficient unto itself to cause cancer, other pathways must be involved such that homeostasis is disrupted.
A pathogenic stimulus causes a reaction, an inflammation. The earliest report on the role of inflammation in carcinogenesis came in 1915 when coal tar derivatives, repeatedly applied to rabbit ears, induced skin cancer [32]. Since then, alkylating agents (sulfur mustard, ethylene dibromide, and many nitrosamines), arsenic, aflatoxins, asbestos, azo dyes, benzene, cigarette smoke, vinyl chloride, and radiation have all been associated with carcinogenesis [18,19,23]. A feature shared by these chemical carcinogens is an extended latency period between repeated exposures and the onset of cancer. A nonchemical example is the chronic inflammation in persistent achalasia associated with an estimated 140-fold increased risk of developing esophageal cancer [33].
The complex, yet insidious, nature of chronic inflammation can be seen both inside and outside the cell. Disruption of the glycocalyx, the thin layer of glycoproteins, and glycolipids that coats nearly every cell, can result in both altered signaling pathways and favor a pro-inflammatory state within the cell [18,19].
One function of the glycocalyx is to protect cells and underlying tissues, while another serves as a direct mechanical-transducing link to couple the guanosine-5′-triphosphate (GTP)-binding proteins of the cell membrane as well as promoting contact with microfilaments inside the cell [18,34]. The regeneration time after disruption of the glycocalyx is about 5–7 days [35]. The consequences of the degradation of the glycocalyx involve increases in the number of intercellular adhesion molecule 1 (ICAM1, cluster of differentiation 54, CD54) on the cell surface, and activates the nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB), thus fostering a pro-inflammatory state marked by elevated leukocyte adhesion [36]. The glycocalyx is found around epithelial, stromal, and tumor cells, and disruption by hyaluronidase and heparinase was reported to protect against flow-regulated invasion [37].
The role of adhesion molecules in carcinogenesis is being increasingly examined. The cytochrome P450 (CYP) metabolized statin, simvastatin, inhibits the adhesion of cancer cells to human mesothelial cells (HMCs) and decreases the adhesion molecules, vascular cell adhesion protein 1 (VCAM-1) on HMCs, and ICAM-1 and the cell surface receptor integrin beta 1 (integrin β1, cluster of differentiation 29, CD29) chain expression in ovarian cancer cells. This opens up the possibility of a therapeutic role for simvastatin in patients with peritoneal carcinomatosis [38] perhaps by applying long-circulating liposomes as a constant delivery system for simvastatin [39].
The experiments by Zeng et al. revealed that blocking metalloproteinases (MMPs), the cell surface glycoprotein cluster of differentiation 44 (CD44), or integrin alpha 3 (integrin α3, cluster of differentiation 49C, CD49C) decreases flow-induced migration. The authors theorized that sphingosine-1-phosphate (S1P), together with glycocalyx, Snail, and MMP signaling, mediate S1P-induced cell transition [40]. The protection of the glycocalyx is induced by S1P, thereby increasing its synthesis on endothelial cells mediated by the phosphatidylinositide 3-kinase (PI3K) pathway [41]. However, upon closer examination, it appears that S1P exerts different effects depending on where it acts within the organism [42, 43, reviewed in 44]. S1P plays a role in the chemotaxis of mast cells to sites of inflammation [45]. The disruption of the molecular homeostasis of sphingosine kinase isoforms (SphK) and S1P is thought to occur during carcinogenesis.
SphK is increased in breast cancer [46,47] and is activated by cytokines, resulting in increases in intracellular S1P [48]. SphK-induced S1P accumulation suppresses apoptosis [49]. Overexpression of SphK facilitates cancer development [45,50]. S1P signaling in Epstein-Barr virus (EBV)-associated nasopharyngeal carcinoma activates protein kinase B (Akt, PKB) and promotes cell migration through S1P receptor 3 (S1PR3), while a knockdown of S1PR3 decreases Akt activation and the S1P-triggered migration of nasopharyngeal carcinoma cells in vitro [51].
In patients with inflammatory bowel disease (IBD), the risk of cancer is increased some 20–30-fold [52]. S1P is thought to be involved in the progression from IBD to cancer [53]. It may be that two homologous proteins (S1PL2021 and S1PL2025), which were recently reported in the pathogenic bacterium Burkholderia pseudomallei K96243, degrade host sphingolipids (SLs) via a mechanism mediated by the pyridoxal 5′-phosphate (PLP)-dependent enzyme S1P lyase (S1PL) [54]. Exosomes secreted by enterobacteria trigger the intestinal epithelium to produce exosome-like nanoparticles derived from mucosa, which contain S1P, chemokine (C-C motif) ligand 20 (CCL20, macrophage inflammatory protein-3, MIP3A), and prostaglandin E2 (PGE2), which, in turn, promote Th17 cells and modulate intestinal inflammation [55].
Other indicators of disruption of cellular homeostasis include catenin delta-1 (protein 120, p120), conjugated bile acids (CBA), and PGE2. The cell adhesion protein 120 (p120, catenin delta-1) expression is elevated in chronic pancreatitis and can be diminished in tumors. It is thought that together with the signaling of extracellular S1P bound to S1P receptor 2 (S1PR2) on neighboring cells these changes could affect the progression to cancer [56]. Furthermore, CBA activate S1PR2 in hepatocytes [57] and, thereby, promote growth in cholangiocarcinoma through activation of S1PR2 [58].
CBA-induced cyclooxygenase 2 (Cox-2) expression in a human HuCCT1 CCA cell line is also associated with S1PR2-mediated upregulation of both Cox-2 expression and PGE2 production, highlighting the role of S1PR2 I bile acid-induced Cox-2 expression and abnormal cell growth [59]. The small molecule 3-(4-chlorophenyl)-adamantane-1-carboxylic acid (pyridin-4-ylmethyl) amide (ABC294640) selectively inhibits sphingosine kinase 2 (SK2) activity leading to dose-dependent decreases in S1P expression [60]. It also targets sphingosine kinase isoforms, sphingosine kinase isoform 1 (SphK1) or sphingosine kinase isoform 2 (SphK2), inhibiting constitutive signal transduction and dose-dependent caspase cleavage, as well as apoptosis for Kaposi's sarcoma-associated herpes virus (KSHV)-positive patient-derived primary effusion lymphoma (PEL) cells [61].
Pathogenic stimulus by virus
Some 2,400 years ago, Hippocrates is believed to have coined the term “herpes” “to describe lesions that appeared to creep or crawl along the skin” although descriptive lesions seem to have been reported on a Summarian Tablet in the third Millenium BC and on the Ebers Papyrus about 1,500 BC [62–65]. Hippocrates (460–377 BC) first described the association of uterine cancer and inflammation [66, reviewed in 67] followed by Galen (approximately 130–210 AD) with “scleroma uteri est tumor subdurus in aliqua parte uteri oxortus qui plerumque ex diuturnis inflammationibus” [68, reviewed in 67]. Arataeus of Cappadocia (approximately first to third century AD) recognized the association of inflammation in liver cancer: “The liver inflammation persists, and the pus remain inside the liver, the pain persists too, the enlargement turns to a rough area and transformed to cancer” (reviewed in Ref. [67]).
John Abruc in 1736 investigated French prostitutes and linked herpes with genital lesions [69]. This was followed by Vidal, who in 1873 associated human herpes virus 1 (HSV-1, HHV-1) associated with oro-facial and human herpes virus 2 (HSV-2, HHV-2) with genital lesions including the suggestion that herpes may be infectious between humans [70], Steiner in 1875 linked human herpes virus 3 (HHV-3, Varizella zoster virus, VZV) with chicken pox [71], and Unna 1883 associated human herpes virus (HSV) with genital lesions but he described herpes as “…one of the most benign of afflictions both to the patient and her public” [72].
Kelsch and Kiener reported two cases of liver cancer in 1876 [73] and the first histological classification was done by Hanot and Gilbert [74]. Virchow differentiated between primary liver cancer and liver metastasis [75] followed by the subsequent distinctions between primary liver cancers derived from hepatocytes resulting in “hepatoma” (later hepatocellular carcinoma or HCC), and “cholangioma” deriving from the intrahepatic bile ducts (later cholangiocellular carcinoma or CCC) [76,77].
The association of liver cancer and fibrosis was pointed out by Sabourin using the term hepatoma [78], and the association of hepatitis involved during liver cancer development was recognized in 1948 [79,80].
The association of hepatitis B virus (HBV) and liver cancer was made by Prince and Sherlock [81,82] and the causal association was accepted [83] while the association of liver fluke and CCC was performed by Kartsurada 1900 and Hou 1955 [84,85, reviewed in 86].
The Irish surgeon, Denis Parsons Burkitt, then in Uganda, discovered that viruses were associated with cancer when he observed swellings in the angles of the jaw in children [87]. In 1961, Burkitt gave a lecture entitled “The Commonest Children's Cancer in Tropical Africa − A Hitherto Unrecognized Syndrome.” In 1964, Michael Anthony Epstein, a pathologist, together with Bert Achong and Yvonne Barr, identified particles of virus in cells cultured from Burkitt's cancer patients [88], demonstrating that viruses were associated with what later became known as Burkitt's lymphoma. In 1968 and 1971, reports appeared that showed that cell-free extracts of human osteosarcomas inoculated in hamsters could induce the mesenchymal cancer, osteosarcoma [89,90]. Later, Balabanova et al. showed that the transformation of normal cells to cancer cells occurred in vitro after infection with two C-type viruses obtained from solid tumors [91,92]. In 1974, Epstein and Kaplan established the SU-DHL-1 lymphoma cell line from a 10-year-old boy suffering from diffuse histiocytic lymphoma [93]. Kaplan et al. later demonstrated in vitro that the SU-DHL-1 virus could infect human hematopoietic cells and transform them to malignant cells [94–96]. Subsequently, Gertrude and Werner Henle with collaborators from their Philadelphia laboratory developed serological markers and antibodies to the offending virus [97]. The first sero-epidemiological studies on EBV were published in the late 1970s. [98]. Henle later showed that EBV could affect a transition of normal lymphocytes to cancerous cells [99].
The association of HSV with chronic inflammation [100–103] and the development of squamous dysplasia, carcinoma in situ, and cervical carcinoma has been proven since the 1860s [104–112]. Since 1981, a herpes classification is available [113]. The Hungarian dermatologist, Moritz Kaposi (1837–1902), reported an idiopathic skin sarcoma [114, reviewed in 115], which was essentially ignored until 1981 when human immunodeficiency virus (HIV) infection and acquired immune deficiency syndrome (AIDS) were associated with Kaposi sarcoma syndrome and opportunistic infections [116]. Human herpes virus 8 (HHV-8) causes PEL, multicentric Castleman disease (MCD), and Karposi sarcoma, a malignant cancer of blood vessels that is common in immune-compromised patients with AIDS [117].
Other viruses have also been causally linked to specific cancers and the association of chronic hepatitis B or C (HBV or HCV) and liver cancer is established [118].
The associations of HPV with cervical carcinoma [119], middle ear carcinoma [120], penile carcinomas [121,122], oropharyngeal cancer [123], tongue cancer [124], esophageal squamous cell carcinoma (ESCC) [125], colorectal cancer (CRC) [126,127], lung cancer [128–130], and breast cancer have since been demonstrated [131].
In regard to the herpes virus, another paradigm is falling. It was propagated for a long time that Alzheimer's disease was caused by mutations in the majority of cases [132–135], although we now know that only some 5% of Alzheimer's cases are causally linked to mutations [136]. Recently, it was pointed out that the majority of cases (≥50%) seem to be caused by chronic inflammation induced by herpes virus species [137].
Pathogenic stimulus by bacteria
The association of Helicobacter pylori and gastric cancer [138] was long ignored [139]. Eventually, the pathogen was shown to cause fibrosis, one step in the proposed six-step sequence of carcinogenesis [18]. It is relevant that >60% of infections by gastric spiral organisms (GSO) were observed in dogs and cats, where they act as a reservoir for subsequent human infection with H. pylori [140]. Dogs infected with this pathogen showed a greater incidence of moderate-to-severe gastritis than dogs that were H. pylori negative [141]. The mode of transmission of this gram-negative bacterium is unclear as is the possibility of transmission via fecal–oral, gastric–oral, oral–oral, and sexual routes. H. pylori was shown to be present and to survive in food samples such as milk, vegetables, and meat, a fact that points to the role of environmental factors in assessing cancer risk [142].
H. pylori uses the serine protease, high temperature requirement A (HtrA), to cleave important components of the tight junctions, such as occluding and claudin-8, and the cell–cell adhesion protein cadherin-1 (E-cadherin), making transcellular migration possible [143]. Bacterial adaptations to the cellular milieu are Type IV secretion systems (T4SSs) found in gram-negative and gram-positive bacteria [144]. In H. pylori infection, T4SSs is formed at the basolateral epithelial membrane. After disrupting cell–cell junctions and binding to the integrin-β1 receptor, the cytotoxin-associated gene A (CagA) protein of H. pylori is injected into the host [143]. It has recently been shown that CagA deregulates autophagy and promotes chronic inflammation through tyrosine-protein kinase Met (c-Met, hepatocyte growth factor receptor, HGFR)-PI3K/Akt- mechanistic target of rapamycin (mTOR) signaling [145].
Disruption of gut bacteria composition as internal pathogenic stimuli
The roles of bacteria as pathogenic stimuli were examined in a population-based study in postmenopausal women by comparing 48 breast cancer cases with 48 contemporaneous age-matched controls [146]. Adjustment for estrogens and other variables showed “…reduced alpha diversity and altered composition of both their IgA-positive- and IgA-negative faecal microbiota,” revealing that “breast cancer cases had significant oestrogen-independent associations with the IgA-positive and IgA-negative gut microbiota.” An investigation of 668 breast tumor tissue and 72 noncancerous adjacent tissue was performed to try to characterize microbiota and to study associations of microbiota with tumor expression profiles [147]. To confirm the presence of microbiota, sequencing of the 16S rRNA gene was used to identify unknown bacteria “or for providing reference identifications for unusual strains” [148]. Thompson et al. found that Escherichia coli was “one of the more prevalent species in the breast tissue and is observed in higher abundance within non-cancer breast tissues.” Furthermore, a greater shift in the abundance of Proteobacteria in cancer tissues was observed compared to Actinobacteria in noncancer tissues. Listeria spp. was associated with expression profiles of genes involved with cell transition and H. influenza was associated with proliferative pathways, such as G2M checkpoint, E2F transcription factors, and mitotic spindle assembly [147].
A nested case-control study of two prospective cohort studies, the American Cancer Society Cancer Prevention Study II Nutrition Cohort (CPS-II) and the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial (PLCO), investigated head and neck squamous cell cancer (HNSCC) and identified 129 incident cases of HNSCC that provided oral mouthwash samples, and that were cancer-free at baseline through bacterial 16S rRNA gene sequencing (oral microbiome composition and bacterial abundances) [149]. Although the statistical power was low, greater abundances of Corynebacterium and Kingella were associated with decreased HNSCC risk. Additionally, there was a tendency for stronger associations with laryngeal cancer and for patients with a tobacco history. Blastocystis sp. was isolated from fecal samples and inoculated into rats treated with the carcinogen, azoxymethane (AOM), Blastocystis sp. co-treatment, and a control group [150]. Histologically, AOM-treated rats with co-administration of Blastocystis sp. cyst resulted in a 1.6-fold increase in the number of crypts compared to AOM-treated rats. Foci of aberrant crypts were observed in AOM-controls and Blastocystis sp. infected AOM-rats. Tubular adenomas with very mild dysplasia were observed in AOM-treated rats compared to major dysplasia with hyperplastic aberrant crypts rats treated with AOM and Blastocystis sp. Furthermore, the authors found higher oxidative stress parameters (lipid peroxidation, protein damage) in Blastocystis sp. infected AOM-rats compared to the AOM-treated controls. Higher levels of advanced oxidative protein products (AOPP) were also found in the serum [150], which is concordant with high plasma AOPP levels found in CRC patients compared to healthy individuals [151].
Methylobacterium radiotolerans was observed to be enriched in breast cancer tissue compared to an abundance of bacterium Sphingomonas yanoikuyae in paired normal tissue [152]. This inverse microbial dysbiosis was concordant to an investigation of both bacteria in sentinel lymph node specimens from breast cancer patients [153]. However, it is not clear how these data explain the observation of 2266 women treated with antibiotics who were found to be at higher risk [2.07 (95% CI = 1.48–2.88)] of developing breast cancer [154].
The role of bacteria as pathogenic stimuli was examined in tissue samples from 57 breast cancer patients and compared to that in breast tissue samples taken from 21 healthy women undergoing cosmetic breast surgery. Bacterial 16S rRNA is amplified from oral, urine rinse and breast tissue collected by surgery, sequenced, and processed. Methylobacterium was found primarily in noncancer patients. Urinary gram-positive bacteria such as corynebacterium, staphylococcus, actinomyces, and Propionibacteriaceae were increased in cancer patients independent of menstrual status. The authors suggested that there was a different local breast cancer microbiome between cancer and noncancer patients [155]. Sequencing of the 16S rRNA gene was used to detect previously unidentified bacteria “or for providing reference identifications for unusual strains” [148]. Wang et al. found that compared to controls, clustered microbiomes in breast cancer tissue, “…driven by a decreased relative abundance of Methylobacterium in cancer patients” [155]. Plant-associated methylobacteria were previously thought to act as coevolved phytosymbionts [156] and methylobacterium rarely causes infections [157], while phytohormones are considered to be protective against cancer, making this observation interesting [158,159].
In another investigation of mucosal tissues from 150 patients who underwent colonoscopy, the authors reported a high degree of concordance between colonization with enterotoxigenic Bacteroides fragilis (ETBF) and low-grade dysplasia (LGD), tubular adenomas (TA), and serrated polyps, and thus with precancerous lesions [160]. Additional evidence for internal pathogenic stimuli comes from the fact that bacterial aggregates are associated with proximal colon cancers [161]. Fusobacterium and Providencia were shown to be associated with the cancer microenvironment [162].
Although dermatophytes usually cause superficial fungal infections of the skin in immunocompromised patients, Trichophyton rubrum can cause deep dermatophytosis with hematogenous dissemination [163]. It has been suggested that dermatophytes could trigger mycosis fungoides [164]. Zhang et al. reported that about 25% of 20 medulloblastomas contained the deoxyribonucleic acid (DNA) of Brucella species by the OMP31 primer/probe set; none of the medulloblastomas had a specific sequence for B. mellitensis, B. suis, or B. abortus [165]. Fusobacterium nucleatum ssp. animalis induced chronic inflammation and monocyte/macrophage activation, which could promote cancer development [166].
Chronic inflammation has been shown to initiate pancreatic carcinogenesis without a K-ras mutation and in the absence of a tumor protein 53 (p53) mutation [167].
Pathogenic stimulus by fluke
CCC has been associated with liver fluke (Opisthorchis viverrini) [168–170]. Thailand has the highest CCC rates worldwide, especially in the northern and northeastern part [171]. This is concordant with the finding that the “...majority of the infected cases (64.3%) were found from the immigrants of northeastern Thailand (the fluke-prevalent region), providing 2.28–2.42 times higher infectious risk on average against the local residents.” The prevalence of O. viverrini, Schistosoma mekongi, and soil-transmitted helminths (STH) is also high in the Lao People's Democratic Republic (Lao PDR), and animal infections rates, as well as water, might serve as a reservoir for humans [172]: “O. viverrini and S. mekongi infection rates among dogs and cats were 25.0% and 14.7%, respectively. Of the cats tested, 53.1% were infected with O. viverrini. Prevalence of O. viverrini and S. mekongi in snails was 0.3% and 0.01%, respectively. Overall prevalence of O. viverrini infection in fresh water fish was 26.9%, with the highest infection rates occurring in Hampala dispa (87.1%).”
According to the six-step carcinogenesis sequence [18], O. viverrini infection is associated with a two to three times increase of plasma inflammatory indicators, 8-isoprostane, malondialdehyde, and nitrate/nitrite, while plasma C-reactive protein (CRP) levels did not differ compared to healthy controls [173]. In a rodent model, inflammation with severe periductal fibrosis and changes in the epithelium of the biliary tract, as pre-cancerous lesions, were induced by the liver fluke Opisthorchis felineus [174]. Furthermore, the combination of O. viverrini infection and chemical carcinogen induced CCC in hamsters through inflammatory mechanisms with an increase of cluster of differentiation 4 positive (CD4+) and interleukin 17 positive (IL-17+) cells, and intense staining of the transcription factor Forkhead-Box-Protein P3 (FoxP3) was reported and was consistent with their protein levels [175]. Applying N-acetyl-5-methoxy tryptamine (melatonin) suppressed IL-17+ inflammatory cells and Foxp3+ cells and increased CD4+ cell infiltration and tumor necrosis factor alpha (TNFα) expression.
IL-17 is a pleiotropic cytokine involved in carcinogenesis [176,177] and possess antitumor effects through a T-cell dependent mechanism [178]. IL-17 reveals heterogeneous immunohistochemistry (IHC) expression in CRC compared to normal tissues [179]. IL-17 is increased in breast cancer, where it enhances TNFα-indeced increase in hypoxia-inducible factor (HIF-1α) with inhibition of vasodilator-stimulated phosphoprotein (VASP) and consequent reduction of adhesion [180]. Activating STAT3 by IL-17 decreases myeloid-derived suppressor cells (MDSCs) in breast cancer [181]. IL-17 stimulates interleukin 6 (IL-6) expression 13-fold and interleukin 8 (IL-8) 28-fold in benign prostate hyperplasia (BPH) cells but in situ measurements showed that the amount of 17mRA in BPH cells did not correlate with IL-6 or IL-8 mRNA but rather was strongly correlated in prostate cancer tissues [182]. Furthermore, IL-17 increases in vivo tumor growth in human nonsmall cell lung cancer (NSCLC) in SCID mice through CXCR-2-dependent angiogenesis [183]. MMP-9, MMP-2 via NF-κB/hypoxia-inducible factor (HIF-1α) pathway is increased by IL-17 in rheumatoid arthritis, promoting cell migration [184], and MMP-9 through mitogen-activated protein kinase 3 (ERK1) and mitogen-activated protein kinase 1 (ERK2) MAPK activation [185].
The IL-1/IL17 signaling axis contributes in inflammatory remodeling of fibrosis in systemic sclerosis [186]. IL-17 increase via IL-6, TNF, CCL20, and C-X-C motif ligand 1 (CXCL1) in heart failure with consecutive enhancing tissue remodeling [187]. The activation of the IL-17/long noncoding RNA-AK081284 (AK081284)/TGFβ1 increases cardiac interstitial fibrosis, while Il-17 knockout results in decrease of remodeled fibrosis and improved cardiac function in mice [188]. There are also contradictory findings, namely, pulmonary fibrosis is increased via IL-17 induced Th17 cells [189], while renal interstitial fibrosis can be inhibited by IL-17 [190], which may be dependent on the IL-17 concentration as low dose IL-17 can prevent diabetic nephropathy [191]. It is known that the effect of IL-17 in allergic airway inflammation depends on the quantity as well as the cellular source of IL-17 [192], and using anti-IL-17 in psoriasis resulted in “dose-dependent reductions from baseline in keratinocyte proliferation, hyperplasia, epidermal thickness, infiltration into the dermis and epidermis by T cells and dendritic cells” [193].
IL-17 seems to have dose- and cell source-dependent inflammatory and pro-fibrotic effect, whereas interleukin 22 (IL-22) was associated with protecting against liver fibrosis through downregulation of the TGF-β1/Notch signaling pathway [194] but being pro-fibrotic in cardiac fibrosis through STAT3/and ERK and IL-17, IL-6, IL-1β, IFN-γ, and TNF-α [195] and pro-atherosclerotic [196]. Both, IL-17 and IL-22 are associated with poor prognosis in liver cancer [197], but the exact roles of IL-17 and IL-22 need to be defined in detail in regard to concentration, kind of inflammation, and period of chronic inflammation involved in signaling axis and cell sources.
New interactions between chronic inflammation contain aquaporines (AQPs) and autophagy, both getting increasingly into cancer research focus [198–200].
Aquaporines (APQ)
The aquaporines (AQPs) are a family of small hydrophobic membrane proteins in the animal and plant kingdom that are involved in transport of water and in water homeostasis, which was first reported in 1992 by Peter Agre's group [201] and for which he received the Nobel Prize in Chemistry in 2003. AQPs are involved in concentrating the urine within the kidneys and the salivary glands as well as within the liver for glycogenesis [202–205].
AQPs are expressed “in many epithelia and endothelia involved in fluid transport, as well as in cell types that are thought not to carry out fluid transport, such as skin, fat and urinary bladder cells” [206]. The various AQP function roles include trans-epithelial fluid transport, tissue swelling, cell migration, fat metabolism, pathophysiology of obesity, immune cell dysfunction, cancer, and other diseases. Piscine aquaporins, in Agnatha (jawless fish), Chondrichthyes (chimaeras, sharks, and rays), Dipnoi (lungfishes), and Teleostei (ray-finned bony fishes) “imply the physiological roles of piscine aquaporins extend at least to osmoregulation, reproduction, and early development” [207]. Research revealed the association of various AQPs with cancer, its development, and metastasis [208–214]. However, AQP seem to be heterogeneously expressed in highly aggressive cancers with increased cell migration and cell proliferation [215].
Papadopoulos et al. proposed a mechanism how AQPs may play a role in a kind of amoeboid cell migration important for metastasis and by which rapid changes in cell volume facilitated by AQP include [216] actin depolymerization and ionic influx with increased osmolality at the front end of the cell, water influx followed by actin re-polymerization with formation of localized cell membrane protrusions. There is hope that AQP inhibitors or modulators might serve as future drugs for slowing down tumor growth and migration [216,217].
The fluid clearance within the pancreatic duct chronic pancreatitis is in addition dependent on AQPs and not just on outflow obstruction: aquaporin 1 (AQP1) is mainly localized at the apical membrane of ductal cells in the human pancreas and AQP1 expression in mice is dependent on the cystic fibrosis transmembrane conductance regulator protein and chlorid channel (CFTR Cl−), channels in mice. Induction of acute pancreatitis by intraperitoneal injection of cerulean in mice resulted in decreased channel expression, which was concordant to findings in human with acute and chronic pancreatitis [218]. AQP1 knockdown worsened the pancreatitis with significant decrease of pancreatic ductal fluid and hydrogencarbonat (HCO3 −) secretion with increased chronic inflammation.
A disruption of AQPs in chronic inflammation and various diseases was observed: upregulation of aquaporin 1 (AQP1) in rheumatoid arthritis [219] versus a downregulation of aquaporin 4 (AQP4) and 8 (AQP8) in colitis-induced mice [220], and increased AQP1, aquaporin 3 (AQP3), aquaporin 7 (AQP7), and aquaporin 8 (AQP8) in early untreated human IBD [221]. The anti-inflammatory mediator, lipoxin A4 (LXA4, 5S,6R,15S-trihydroxy-7E,9E,11Z,13E-eicosatetraenoic acid) [222], regulates aquaporin 5 (AQP5) and metalloproteinase 9 (MMP-9, gelatinase B) in pancreatitis-associated lung injury through the protein kinase C (PKC)/ src-suppressed C-kinase substrate (SSeCKS) pathway with F-actin reconstruction [223].
AQP1 is thought to be involved in the differentiation of gastric cancer cells [224], and AQP3 and aquaporine 9 (AQP9) can be induced by arsenite with inhibition of apoptosis via decreasing p53 and increasing B-cell lymphoma 2 (Bcl-2) [225]. AQP1 and AQP5 are increasingly expressed at the apical membrane of intercalated pancreatic duct cells [226], and increased AQP5 levels result into “retinoblastoma protein phosphorylation through the formation of a nuclear complex with cyclin D1 and CDK4” [227]. AQP3 was found to be increased in advanced stages of pancreatic cancer, and AQP5 is more closely associated with tumor differentiation [228].
Autophagy
The mechanism a cell uses to rid itself of unnecessary or dysfunctional components is termed autophagy [229]. It was coined by Christian de Duve in 1963 [230, reviewed in 229]. Intracellular bacterial infections [231] such as propionibacterium acne [232], Shigella and Salmonella [233], H. pylori [234], and viruses [235] can generate autophagy. The accumulation of intracellular amino acids induces a dissociation of mTOR from the membrane with downregulation of its activity. At the same time, the amino acids activate EIF2AK4/GCN2-EIF2S1/eIF2α/ATF-3 signaling (eukaryotic translation initiation factor 2-alpha kinase 4 (EIF2AK4)/general control nonderepressible 2 (Gcn2)-eukaryotic translation initiation factor 2 subunit 1 (eIF2α, EIF2S1)/eukaryotic translation initiation factor 2 subunit 1 (eIF2α)/cyclic AMP-dependent transcription factor (ATF-3)) [233].
It has recently been reported that endogenous nuclear fragments, such as chromatin, evoke a chronic inflammatory response within the cytoplasm also leading to autophagy [236]. An external pathogenic stimulus also induces an internal protection shield process for the cell, termed consecutive autophagy [237].
Autophagy is linked to subclinical inflammation especially in precancerous lesions such as inflammatory bowel diseases (IBD), in which autophagy controls colitis by decreasing TNF-induced apoptosis [238], which is why autophagy in IBD is seen as a protection against cell suicide for the gut epithelium [239]. But the contradictory findings have not been fully explained. Chronic pancreatitis is associated with progressive chronic inflammation and a precancerous condition for pancreatic cancer [240,241].
Long noncoding RNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is increased in pancreatic cancer and is associated with poor survival [242,243] and promotes cancer progression by autophagy activation [244]. Retinoblastoma-coiled coil protein 1 (RB1CC1) mediates autophagy, which is seen as a key event for the activation of pancreatic stellate cells (PSCs), and knocking down retinoblastoma-coiled coil protein 1 (RB1CC1) expression alleviates autophagy, which suggests an opportunity of a potential use as a therapeutic approach in chronic pancreatitis and fibrosis [245].
On the one hand, site lipopolysaccharide (LPS)-induced autophagy is associated to promote autophagy in gingival fibroblasts [246], while Mycobacterium leprae can inhibit autophagy [247]. Autophagy may be even a protection strategy of cancer cells against multimodal therapy-induced apoptosis [248]. As pointed out, there are gaps in our knowledge in regard to dysregulation of autophagy [249].
Chronic inflammation
The interactions between mast cells, T-cells, neutrophils, granulocytes, and macrophages are complex such that the chronic activation of one or more of these results in the release of pro-inflammatory cytokines (examples included in Fig. 1) and those, in turn, are linked to the development of fibrosis [250–253]. The chronic activation of macrophages brings about a chronic release of TNFα, interleukin beta 1 (IL-β1), and oncostatin-M (OSM) and leads to a continuous activation of fibroblasts [254–256]. Chronic activation of mast cells also produces a release of cytokines as well as activating T-cells and fibroblasts [257] (Fig. 1). T-cells, involving a ligand such as cluster of differentiation 40 (CD40) in antigen presenting cells, mediate the contact activation of endothelial cells [258]. Such a process is one plausible mechanism to generate the observed chronic inflammation. Post-partum-activated fibroblasts were associated with increased Ly6C+ monocytes, decreased CD8+ T cells, and apoptosis. They displayed pro-tumorigenic, immunosuppressive activity through Cox-2/PGE2-dependent pathways [259]. This phenomenon is also a reason to consider immunosuppression in cancer therapy.
The immunosuppressive enzyme, indoleamine-2,3-dioxygenase (IDO), is produced in all tissue types [260]. Mice cells transfected with IDO become resistant to immunologic rejection [261], and IDO expression is associated with various cancers such as pancreatic [262,263], gastric [264,265], colorectal [266], breast [267], ovarian, and endometrial [268,269]. The expression of IDO isoforms differs among the various cells; tryptophan degradation by IDO is effected entirely by IDO1 but not IDO2 [264]. Applications of the heme precursor compound, zinc protoporphyrin IX (ZnPP), in a preclinical melanoma model, effectively inhibited IDO [270].
Chronic fibroblast activation alone can lead to a vicious cycle as seen in liver cirrhosis, the end stage of chronic hepatitis. It is a tissue with an abnormal reconstruction of the lobular architecture that contains parenchymal nodules and septa that link the portal with central canals [271]. Popper described the vicious cycle in the creation of hepatocellular fibrosis: “…hepatocellular injury and the associated inflammation produce pericellular fibrosis interfering with cellular nutrition and septa-disturbing hemodynamics. This, in turn, induces additional liver cell injury.”
The negative regulator of Wnt signaling [272], the tumor-suppressor gene, adenomatous polyposis coli (APC), exhibits multiple functions. Silenced APC by promoter methylation was greater in HCC tissue than in noncancerous tissue and the level of APC protein expression was reduced in HCC [273]. The APC tumor suppressor gene also downregulates intestinal transport mediated by both electrogenic sodium–glucose transport protein 1 (SGLT1) and by the Na+/H+ exchanger (NHE3) [274]. It increases the nuclear factor of activated T cells (NFAT) in a microtubule-dependent fashion. APC deficiency in mice reduced NFAT within the nucleus of intestinal regulatory T cells (Tregs), leaving a disturbed Treg differentiation together with anti-inflammatory cytokine, interleukin 10 (IL-10) [275]. APC migration in prostate cancer cells is induced by transforming growth factor beta 1 (TGF-β1) and regulated by Smad7 and p38 mitogen-activated protein kinase (p38 MAPK) [276]. The interaction with microtubules plays an important role in this process.
Aspirin has been demonstrated to inhibit nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling and increase APC expression. Furthermore, it is associated with decreased polyposis in mice as well as in familial adenomatous polyposis (FAP) patients, and decreases the risk of developing CRC [277, reviewed in 278].
For the past 25 years, APC was thought to serve as one of the models for the origin of CRC as the APC mutation is also associated with both familial adenomatous polyposis (FAP) [279] and CRC [280]. However, APC mutations are not seen in mice models that were attributed to the limited lifespan of mice [281]. However, a few years ago it was shown that APC mutations occur at a late stage of tumor formation [282]. From this, and the observation that in a recent mouse model with a germline APC mutation, ApcmNLS/mNLS mice rarely developed tumors [283], the hysteron proteron syllogism should be noted “…because the first event (mutations), in fact occur later in the process, i.e., only after the cell has been transformed from a normal cell to a cancer cell via a process termed carcinogenesis” [23].
Nuclear APC protects against colitis-associated colon tumorigenesis by suppressing inflammation, inhibiting Wnt signaling, and increasing colonocyte differentiation and mucus production [284]. Promoter methylation and APC silencing (reviewed in Refs. [285,286]) were observed in about 70% of inflammatory breast cancers [287], as well as in some 90% of lung cancers [288,289] (and prostate cancers [290–292]). This rate was equaled in both sporadic and familial breast cancer [293], again questioning the role of mutations as being causal to the majority of cancers.
Epithelial cancer cells use IL-β1 as a communication factor to instruct stromal fibroblasts, through p53, to generate a pro-tumorigenic inflammatory microenvironment. This process is mediated (without any mutations) through p53/NF-κB and results in chronic inflammation and fibroblast activation in ovarian cancer [294]. Anti-inflammatory therapy with Ustekinumab, a p40 subunit inhibitor of interleukin 12 (IL-12) and interleukin 23 (IL-23), in patients suffering from Crohn's disease, who were intolerant to TNF antagonist therapy, yielded positive laboratory and endoscopic responses [295]. Both findings suggest that yet chronic inflammation is an early and obligatory event in the genesis of cancer.
The activation and interaction of T cells with neutrophils and the associated cytokine release result in a continuous activation of macrophages, which, in turn, release metalloproteinase 3 (MMP-3, stromelysin-1). MMP-3 activates Pro-MMP1 and Pro-MMP-7 [296,297] (Fig. 1). Metalloproteinase 1 (MMP-1, interstitial collagenase) results in degradation of the extracellular matrix (ECM) with an increase of gelatin, metalloproteinase 7 (MMP-7), and elastin [298–301]. Furthermore, Snail and Smad interacting protein 1 (SIP1) are associated with an upregulation of the MMP family − MMP-1, metalloproteinase 2 (MMP-2, gelatinase A), MMP-7, and metalloproteinase 14 (MMP-14) − together with a downregulation of E-Cadherin [302,303]. The resultant effect increases remodeling of the microenvironment favoring the development of the precancerous niche (PCN) while also downregulating apoptosis.
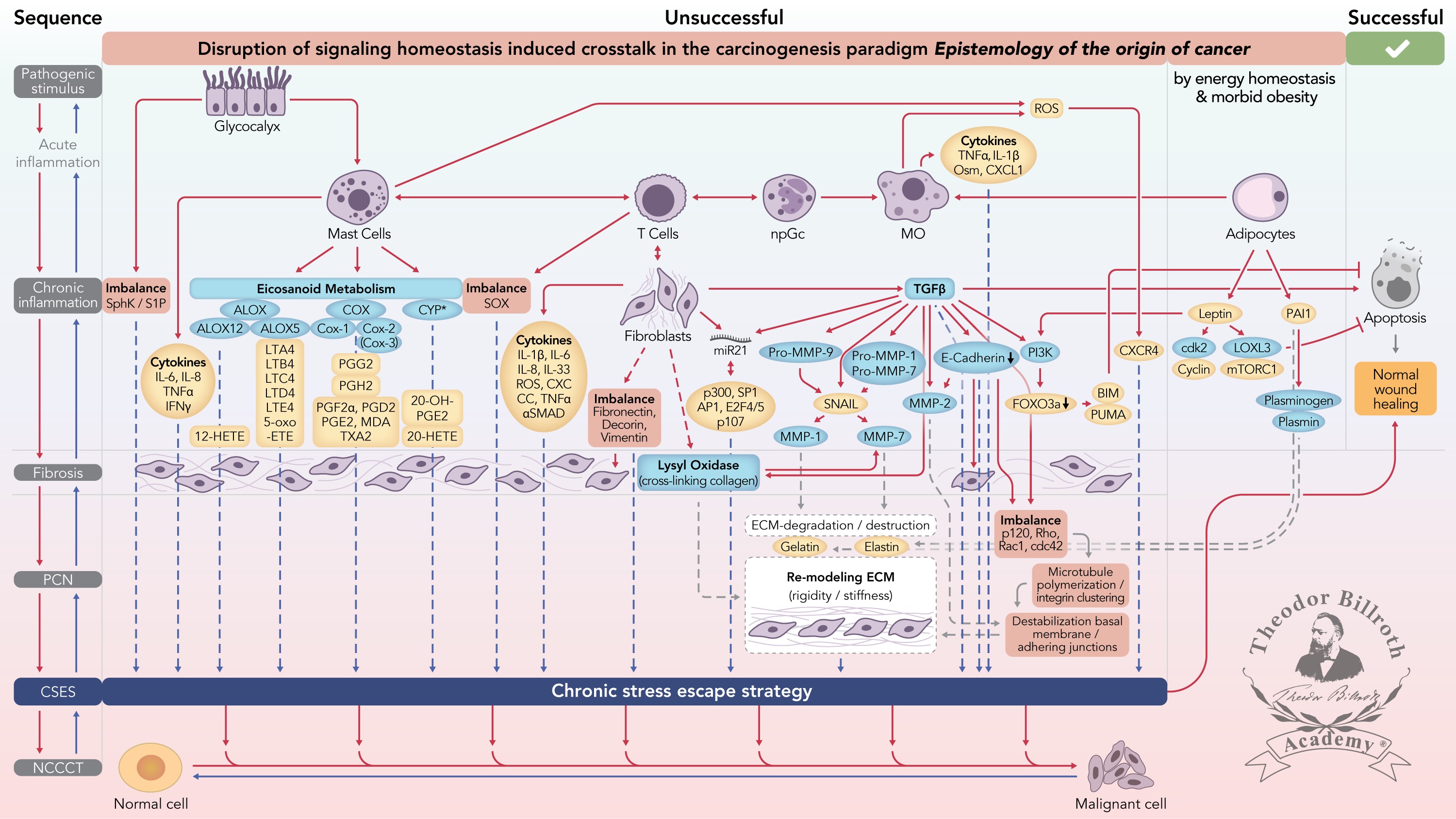
Figure 1 Simplified scheme of the disruption of signaling homeostasis-induced crosstalk in the carcinogenesis paradigm “epistemology of the origin of cancer” consisting of a six-step sequence: (1) a pathogenic stimulus followed by (2) chronic inflammation from which develops (3) fibrosis with associated remodeling of the cellular microenvironment; and from these changes a (4) precancerous niche (PCN), a product of fibrosis, with remodeling by persistent inflammation, develops that triggers the deployment of (5) a chronic stress escape strategy and when this fails resolve it by (6) normal cell to cancerous cell transition (NCCCT) by PCN-induced cell matrix stress occurs. This figure was published as original illustration in paper 3 of this Special Issue – Disruption of homeostasis-induced signaling and crosstalk in the carcinogenesis paradigm “Epistemology of the origin of cancer” entitled “Chronic inflammation evoked by pathogenic stimulus during carcinogenesis”. We point out, that to the complexity of the content of the Special Issue the original and/or modified version of the original illustration was republished within the following papers of the Special Issue: paper 5 “Microbiome and morbid obesity increase pathogenic stimulus diversity”, paper 6 “Precancerous niche (PCN), a product of fibrosis with remodeling by incessant chronic inflammation”, paper 7 “Metformin alters signaling homeostasis”, paper 8 “Transition from normal to cancerous cell by precancerous niche (PCN) induced chronic cell-matrix stress” and paper 9 “NF-κB signaling and crosstalk during carcinogenesis”. Nomenclature: The nomenclature common abbreviations are in bold, followed by the common trivial names (if available) and (if available) by the name in accordance to the International Union of Pure and Applied Chemistry (IUPAC): PCN precancerous niche; CSES chronic stress escape strategy; NCCCT normal cell to cancerous cell transition; SphK sphingosine kinase isoform; S1P sphingosine-1-phosphate; IL-6 interleukin 6; IL-8 interleukin 8; TNFα tumor necrosis factor alpha; IFNγ interferon gamma; ALOX lipoxygenase, arachidonate lipoxygenase; ALOX12 12-lipoxygenase, 12-LOX, 12S-LOX, arachidonate 12-lipoxygenase 12S type; ALOX5 5-lipoxygenase, 5-LOX, arachidonate 5-lipoxygenase; 12-HETE 12-hydroxyeicosatetraenoic acid; LTA4 leukotriene A4, 4-[(2S,3S)-3-[(1E,3E,5Z,8Z)-tetradeca-1,3,5,8-tetraenyl]oxiran-2-yl]butanoic acid; LTB4 leukotriene B4, (5S,6Z,8E,10E,12R,14Z)-5,12-dihydroxyicosa-6,8,10,14-tetraenoic acid; LTC4 leukotriene C4, (5S,6R,7E,9E,11Z,14Z)-6-[(2R)-2-[[(4S)-4-amino-4-carboxybutanoyl]amino]-3-(carboxymethylamino)-3-oxopropyl]sulfanyl-5-hydroxyicosa-7,9,11,14-tetraenoic acid; LTD4 leukotriene D4, (5S,6R,7E,9E,11Z,14Z)-6-[(2R)-2-amino-3-(carboxymethylamino)-3-oxopropyl]sulfanyl-5-hydroxyicosa-7,9,11,14-tetraenoic acid; LTE4 leukotriene E4, (5S,6R,7E,9E,11Z,14Z)-6-[(2R)-2-amino-2-carboxyethyl]sulfanyl-5-hydroxyicosa-7,9,11,14-tetraenoic acid; 5-oxo-ETE (6E,8Z,11Z,14Z)-5-oxoicosa-6,8,11,14-tetraenoic acid; Cox cyclooxygenase; Cox-1 cyclooxygenase 1; Cox-2 cyclooxygenase 2; Cox-3 isoform of Cox-2 (therefore in brakes); PGG2 prostaglandin G2, (Z)-7-[(1S,4R,5R,6R)-5-[(E,3S)-3-hydroperoxyoct-1-enyl]-2,3-dioxabicyclo[2.2.1]heptan-6-yl]hept-5-enoic acid; PGH2 prostaglandin H2, (Z)-7-[(1S,4R,5R,6R)-5-[(E,3S)-3-hydroxyoct-1-enyl]-2,3-dioxabicyclo[2.2.1]heptan-6-yl]hept-5-enoic acid; PGFF2α prostaglandine F2 alpha, (Z)-7-[(1R,2R,3R,5S)-3,5-dihydroxy-2-[(E,3S)-3-hydroxyoct-1-enyl]cyclopentyl]hept-5-enoic acid; PGD2 prostaglandin D2, (Z)-7-[(1R,2R,5S)-5-hydroxy-2-[(E,3S)-3-hydroxyoct-1-enyl]-3-oxocyclopentyl]hept-5-enoic acid; PGE2 prostaglandin E2, (Z)-7-[(1R,2R,3R)-3-hydroxy-2-[(E,3S)-3-hydroxyoct-1-enyl]-5-oxocyclopentyl]hept-5-enoic acid; MDA malondialdehyde, propanedial; TXA2 thromboxane A2, (Z)-7-[(1S,2S,3R,5S)-3-[(E,3S)-3-hydroxyoct-1-enyl]-4,6-dioxabicyclo[3.1.1]heptan-2-yl]hept-5-enoic acid; CYP* cytochrome P450 isoforms; 20-OH-PGE2 20-hydroxy prostaglandin E2; 20-HETE 20-hydroxyeicosatetraenoic acid, (5Z,8Z,11Z,14Z)-20-hydroxyicosa-5,8,11,14-tetraenoic acid; SOX [sex-determining region Y (Sry) box-containing] transcription factor family; IL-β1 interleukin beta 1; IL-33 interleukin 33; ROS reactive oxygen species; CXC CC chemokine receptors; αSMAD alpha-smooth muscle actin; miR21 micro RNA-21; p300 protein 300 (p300-CBP coactivator family); SP1 specificity protein 1; AP1 activator protein 1; E2F4/5 cytoplasmic complex of Smad3, retinoblastoma-like protein 1 (P107, RBL1), E2F4/5 and d-prostanoid (DP1); p107 retinoblastoma-like protein 1, RBL1; TGFβ transforming growth factor beta; Pro-MMP-9 pro-matrix metalloproteinase 9; Pro-MMP-1 pro-matrix metalloproteinase 1; Pro-MMP-7 pro-matrix metalloproteinase 7; SNAIL zinc finger protein SNAI1; MMP-1 matrix metalloproteinase 1; MMP-7 matrix metalloproteinase 7; MMP-2 matrix metalloproteinase 2; E-Cadherin CAM 120/80 or epithelial cadherin, cadherin-1, epithelial cadherin; CXCL1 chemokine (C–X–C motif) ligand 1; Osm oncostatin-M; PI3K phosphatidylinositide 3-kinase; FOXO3a forkhead box protein O3a; p120 catenin delta-1, protein 120; Rho Ras homolog gene family, member A; Rac1 Ras-related C3 botulinum toxin substrate 1; cdc42 cell division control protein 42 homolog; BIM Bcl-2 interacting mediator of cell death; PUMA BH3-only protein; CXCR4 C–X–C motif of chemokine receptor 4; cdk2 cyclin-dependent kinase 2;LOXL3 lysyl oxidase homolog 3; mTORc1 rapamycin complex 1; PAI1 plasminogen activator inhibitor-1.
Summary
Unresolved chronic inflammation is an early and necessary phenomenon in the six-step carcinogenesis paradigm. The signaling and crosstalk pathways that go awry during chronic inflammation are complex because they occur both inside and outside the cell and include various cell types, proteins, and proinflammatory cytokines [139,222,304]. Even intracellular nuclear fragments, such as chromatin, trigger chronic inflammation and affect autophagy. One end product of persistent inflammation is fibrosis, which is a preliminary stage before the formation of the PCN [139]. Various pathogenic stimuli associated with different neoplasms result in persistent inflammatory micromilieu inducing an ongoing dysregulation of homeostasis at the cell, tissue, and organ levels. This disruption of homeostasis is an obligatory early step in carcinogenesis. Similar to other steps in carcinogenesis, a mutation is not necessary to explain carcinogenesis. The interplay of various cells, such as mast cells, macrophages, monocytes, T cells, and fibroblasts with diverse cytokines reveals considerable complexity. The “Disruption of signaling homeostasis induced crosstalk in the carcinogenesis paradigm Epistemology of the origin of cancer” (Fig. 1) becomes more complex by the involvement of the microbiome and obesity [305] and various hydrophobic hormone-like substances, built out of polyunsaturated fatty acids: the eicosanoids [222].
Nomenclature of abbreviations (used in the text and the figure)
5-oxo-ETE: (6E,8Z,11Z,14Z)-5-oxoicosa-6,8,11,14-tetraenoic acid
12-HETE: 12-hydroxyeicosatetraenoic acid
20-OH-PGE2: 20-hydroxy prostaglandin E2
20-HETE: 20-hydroxyeicosatetraenoic acid, (5Z,8Z,11Z,14Z)-20-hydroxyicosa-5,8,11,14-tetraenoic acid
αSMAD: alpha-smooth muscle actin
ABC294640: small-molecule 3-(4-chlorophenyl)-adamantane-1-carboxylic acid (pyridin-4-ylmethyl) amide sphingosine kinase (SK) inhibitor
AK081284: long noncoding RNA-AK081284
AIDS: acquired immune deficiency syndrome
Akt: protein kinase B (PKB)
ALOX: lipoxygenase, arachidonate lipoxygenase
ALOX12: 12-lipoxygenase, 12-LOX, 12S-LOX, arachidonate 12-lipoxygenase 12S type
ALOX5: 5-lipoxygenase, 5-LOX, arachidonate 5-lipoxygenase
AP1: activator protein 1
APC: adenomatous polyposis coli
AOM: azoxymethane
AOPP: advanced oxidative protein products
AQP: aquaporines
AQP1: aquaporine 1
AQP3: aquaporine 3
AQP4: aquaporine 4
AQP5: aquaporine 5
AQP7: aquaporine 7
AQP8: aquaporine 8
AQP9: aquaporine 9
ATF-3: cyclic AMP-dependent transcription factor
AT-Cox2: aspirin-triggered cyclooxygenase 2
Bcl-2: B-cell lymphoma 2
BIM: Bcl-2 interacting mediator of cell death
BPH: benign prostate hyperplasia
CFTR Cl: cystic fibrosis transmembrane conductance regulator (CFTR) and chloride channel (Cl−)
c-c-motif: chemokine
cadherin-1: E-cadherin
CagA: cytotoxin-associated gene A
CBA: conjugated bile acid
CCC: cholangiocellular carcinoma
CCL20: chemokine (C–C motif) ligand 20, macrophage inflammatory protein-3 (MIP3A)
CD4+: cluster of differentiation 4 positive
CD40: cluster of differentiation 40
CD44: cluster of differentiation 44
CD49C: cluster of differentiation 49C (integrin alpha 3, integrin α3)
CD54: cluster of differentiation 54 (intercellular adhesion molecule 1, ICAM-1)
cdc42: cell division control protein 42 homolog
cdk2: cyclin-dependent kinase 2
c-Met: tyrosine-protein kinase Met (hepatocyte growth factor receptor, HGFR)
Cox: cyclooxygenase
Cox-1: cyclooxygenase 1
Cox-2: cyclooxygenase 2
(Cox-3): cyclooxygenase 2 isoform (Cox-2 isoform)
CPS-II: Cancer Prevention Study II by the American Cancer Society
CRC: colorectal cancer
CRP: C-reactive protein
CSES: chronic stress escape strategy
CXC CC: chemokine receptors
CXCL1: chemokine (C–X–C motif) ligand 1
CXCR4: C–X–C motif of chemokine receptor 4
CYP: cytochrome P450
CYP*: cytochrome P450 isoforms
DNA: deoxyribonucleic acid
E2F4/5: cytoplasmic complex of Smad3, retinoblastoma-like protein 1 (P107, RBL1), E2F4/5 and d-prostanoid (DP1)
EBV: Epstein–Barr virus
E-cadherin: CAM 120/80 or epithelial cadherin, cadherin-1, epithelial cadherin
ECM: extracellular matrix
EIF2AK4 : eukaryotic translation initiation factor 2-alpha kinase 4
eIF2α: eukaryotic translation initiation factor 2 subunit alpha (EIF2S1)
EIF2S1: eukaryotic translation initiation factor 2 subunit alpha (eIF2α)
ERK1: mitogen-activated protein kinase 3
ERK2: mitogen-activated protein kinase 1
ESCC: esophageal squamous cell carcinoma
ETBF: enterotoxigenic Bacteroides fragilis
FAP: familial adenomatous polyposis
FOXO3a: forkhead box protein O3a
FoxP3: forkhead box protein P3
Gcn2: general control nonderepressible 2
GSO: gastric spiral organisms
GTP: guanosine-5′-triphosphate
HBV: hepatitis B virus
HCC: hepatocellular carcinoma
HCO3- : hydrogen carbonate
HCV: hepatitis C virus
HGFR: hepatocyte growth factor receptor (tyrosine-protein kinase Met, c-Met)
HHV-1: human herpes virus 1 (HSV-1)
HHV-2: human herpes virus 2 (HSV-2)
HHV-3: human herpes virus 3 (varizella zoster virus, VZV)
HHV-8: human herpes virus 8, Kaposi's sarcoma-associated herpes virus (KSHV)
HIF-1α: hypoxia-inducible factor
HIV: human immunodeficiency virus
HMC: human mesothelial cells
HNSCC: head and neck squamous cell cancer
HPV: human papilloma virus
HSV: human herpes virus
HSV-1: human herpes virus 1 (HHV-1)
HSV-2: human herpes virus 2 (HHV-2)
HtrA: high temperature requirement A (serin protease)
IBD: inflammatory bowel disease
ICAM-1: intercellular adhesion molecule 1 (cluster of differentiation 54, CD54)
IDO: indoleamine-2,3-dioxygenase
IFNγ: interferon gamma
IHC: immunohistochemistry
IL-β1: interleukin beta 1
IL-6: interleukin 6
IL-8: interleukin 8
IL-10: interleukin 10
IL-12: interleukin 12
IL-17: interleukin 17
IL-17+: interleukin 17 positive
IL-22: interleukin 22
IL-23: interleukin 23
IL-33: interleukin 33
Integrin α3: integrin alpha 3 (cluster of differentiation 49C, CD49C)
Integrin β1: integrin beta 1 (cluster of differentiation 29, CD29)
KSHV: Kaposi's sarcoma-associated herpes virus, human herpes virus 8 (HHV-8)
LGD: low-grade dysplasia
LPS: lipopolysaccharide
LOXL3: lysyl oxidase homolog 3
LTA4: leukotriene A4, 4-[(2S,3S)-3-[(1E,3E,5Z,8Z)-tetradeca-1,3,5,8-tetraenyl]oxiran-2-yl]butanoic acid
LTB4: leukotriene B4, (5S,6Z,8E,10E,12R,14Z)-5,12-dihydroxyicosa-6,8,10,14-tetraenoic acid
LTC4: leukotriene C4, (5S,6R,7E,9E,11Z,14Z)-6-[(2R)-2-[[(4S)-4-amino-4-carboxybutanoyl]amino]-3-(carboxymethylamino)-3-oxopropyl]sulfanyl-5-hydroxyicosa-7,9,11,14-tetraenoic acid
LTD4: leukotriene D4, (5S,6R,7E,9E,11Z,14Z)-6-[(2R)-2-amino-3-(carboxymethylamino)-3-oxopropyl]sulfanyl-5-hydroxyicosa-7,9,11,14-tetraenoic acid
LTE4: leukotriene E4, (5S,6R,7E,9E,11Z,14Z)-6-[(2R)-2-amino-2-carboxyethyl]sulfanyl-5-hydroxyicosa-7,9,11,14-tetraenoic acid
LXA4: lipoxin A4, 5S,6R,15S-trihydroxy-7E,9E,11Z,13E-eicosatetraenoic acid
MALAT1: metastasis-associated lung adenocarcinoma transcript 1
MAPK: mitogen-activated protein kinase (see ERK1 and ERK2)
MCD: multicentric Castleman disease
MDA: malondialdehyde, propanedial
MDSC: myeloid-derived suppressor cell
MIP3A: macrophage inflammatory protein-3, chemokine (C–C motif) ligand 20 (CCL20)
miR21:
